Improving mRNA’s stability and immunogenicity with an algorithm tool
Written by Jui-Lin Chen, Ph.D. and Chia-Kuei (Simon) Mo

Pfizer-BioNTech COVID-19 mRNA vaccine. Image from Unsplash.
During the pandemic of COVID-19, mRNA vaccine has been added as a powerful vaccine platform to the arsenal against SARS-CoV-2, the virus that causes COVID-19, and other emerging pathogens. Yet, mRNA’s chemical instability is one of the biggest shortcomings of this novel vaccine platform. A research team led by Liang Huang at the California division of Baidu Research developed an algorithm tool to optimize the sequence of mRNA for the COVID-19 vaccine, leading to improved stability and the vaccine-induced immune response.
In January 2020, the first case of COVID-19 was confirmed in the U.S., and on December 11th and 18th of the same year, U.S. FDA approved the mRNA vaccines developed by Pfizer-BioNTech and Moderna for emergency use. We were all awed by the fact that these companies can readily develop a vaccine for a new virus within a year. Normally, a vaccine can take years from development to FDA approval. Part of the reason is that, unlike traditional vaccines, mRNA vaccines can be synthesized in large quantities without live cells. Traditional subunit vaccines such as HPV and flu vaccines are produced in live cell cultures and live eggs, respectively. Such production and purification processes can be very time-consuming.
And how do mRNA vaccines work? No matter what the platform is the function of all the vaccines is the same — presenting foreign antigens to the immune system (also see “A Brief History of Vaccines: an endless war between humans and pathogens”). Traditional vaccines induce immunity by presenting the antigen itself. The antigen can be a protein such as the SARS-CoV-2 Spike protein in Novavax’s COVID-19 vaccine, or a polysaccharide like in the pneumococcal polysaccharide vaccine (PPSV23) for pneumococcal bacteria. Instead of presenting the antigen to the immune cells, mRNA vaccines provide the recipe for the cells to produce the antigen themselves. For example, upon the immunization of a COVID-19 mRNA vaccine, the vaccinee’s cells are given the recipe to produce the SARS-CoV-2 Spike protein and then they present the Spike protein as an antigen to the immune system.
However, compared to protein-based vaccines, mRNA is less stable. mRNA molecules are more susceptible to oxidation and alkaline environment, and a class of enzymes called RNases that are ubiquitous in the body fluid can quickly break down mRNA molecules. Therefore, vaccines from Pfizer-BioNTech and Moderna are delivered by lipid nanoparticles to protect mRNA from degradation. Even delivered by lipid nanoparticle technology, mRNA vaccines are recommended to be stored between -90°C and -60°C (-130°F and -76°F) as guided by the U.S. CDC, and their shelf life in the refrigerator is only 10 weeks. Such a stringent storage condition makes the distribution of mRNA vaccine challenging especially in middle- and low-income areas, where cold-chain is not well established.
Each RNA sequence can fold in a unique way, like ribbons, two ends of the RNA can fold and pair together to form a double-stranded structure and some regions can remain single-stranded. This unique folded structure is known as a secondary structure. A study shows that double-stranded RNA is far less susceptible to alkaline hydrolysis — the hydrolysis rate of double-stranded RNA is 13.8 times slower than single-stranded RNA. That being said, one can increase the stability of the mRNA vaccines by optimizing the structure of mRNA. However, it’s easy to say than done. RNAs are composed of 4 different nucleotides (A, U, C, G), and 3 consecutive nucleotides form a codon (64 combinatorial possibilities) that determines which one of the 20 amino acids is used, and then the chain of amino acids forms a protein. Therefore, the bigger the protein is, the more possible sequences the encoding mRNA can have. Using SARS-CoV-2 Spike protein as an example, it has 1,273 amino acids and can be encoded by ~2.4×10^632 mRNA sequences! The astronomical number of possible sequences poses an insurmountable computational challenge.

The process of hydrolysis of the phosphodiester bonds in RNA molecules (Left). Double-stranded RNA (dsRNA) is less susceptible to hydrolysis and has much longer half-life than single stranded RNA (ssRNA). Image from Environmental Science&Technology by Ke Zhang et al.
To solve this issue, a research team led by Dr. Liang Huang in California developed an algorithm tool named LinearDesign. LinearDesign predicts the mRNA secondary structure by using concepts and techniques in computational linguistics, a field study method that enables computers to process natural human language. The main idea of LinearDesign is to view the problem of finding the optimal secondary structure of mRNA as finding the most probable sentence that matches a given sound. For instance, when a person says ”How's the weather tomorrow?” it could sound like “House the weather tomorrow?”, or “ How’s the whether to more row?”. The computer then uses some rules of natural language, scores each possible interpretation, and selects the one that makes the most sense. Similarly, LinearDesign uses known mRNA sequences and structures to evaluate and score mRNA sequences to find candidates with the highest stability. Another advantage of LinearDesign is that it also takes into account codon optimization (i.e., choosing codons that are more common in human cells) and achieves joint optimization to find an mRNA sequence that has both high stability and high expression efficiency in human cells.
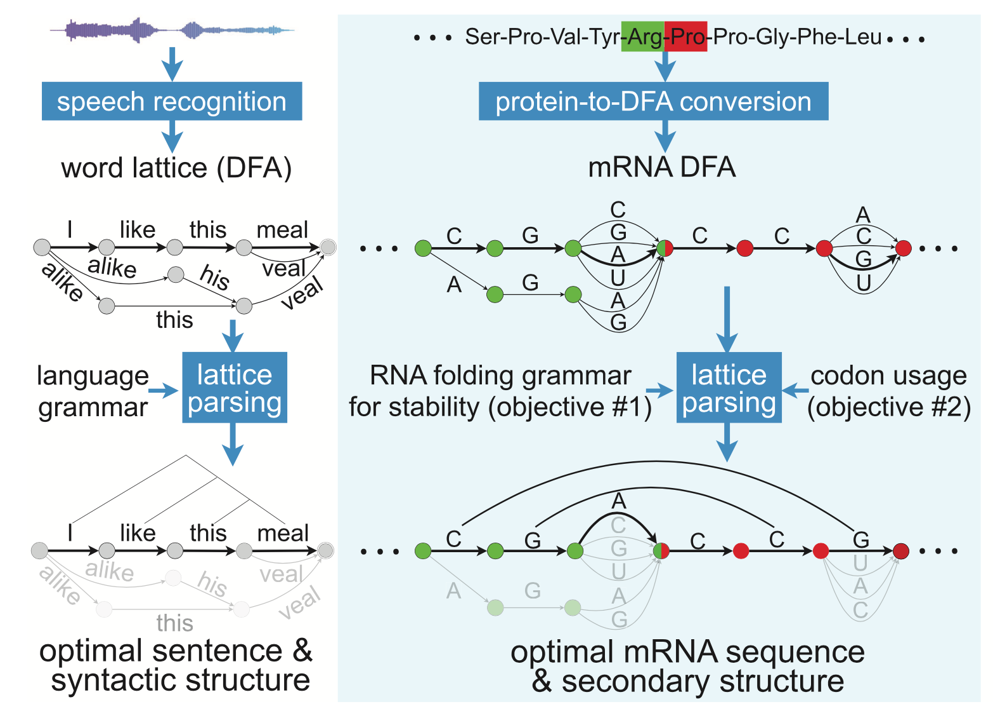
LinearDesign applies concepts from speech recognition to identify optimal mRNA sequences. Image from Fig1C of the LinearDesign manuscript by Liang Huang et al in Nature.
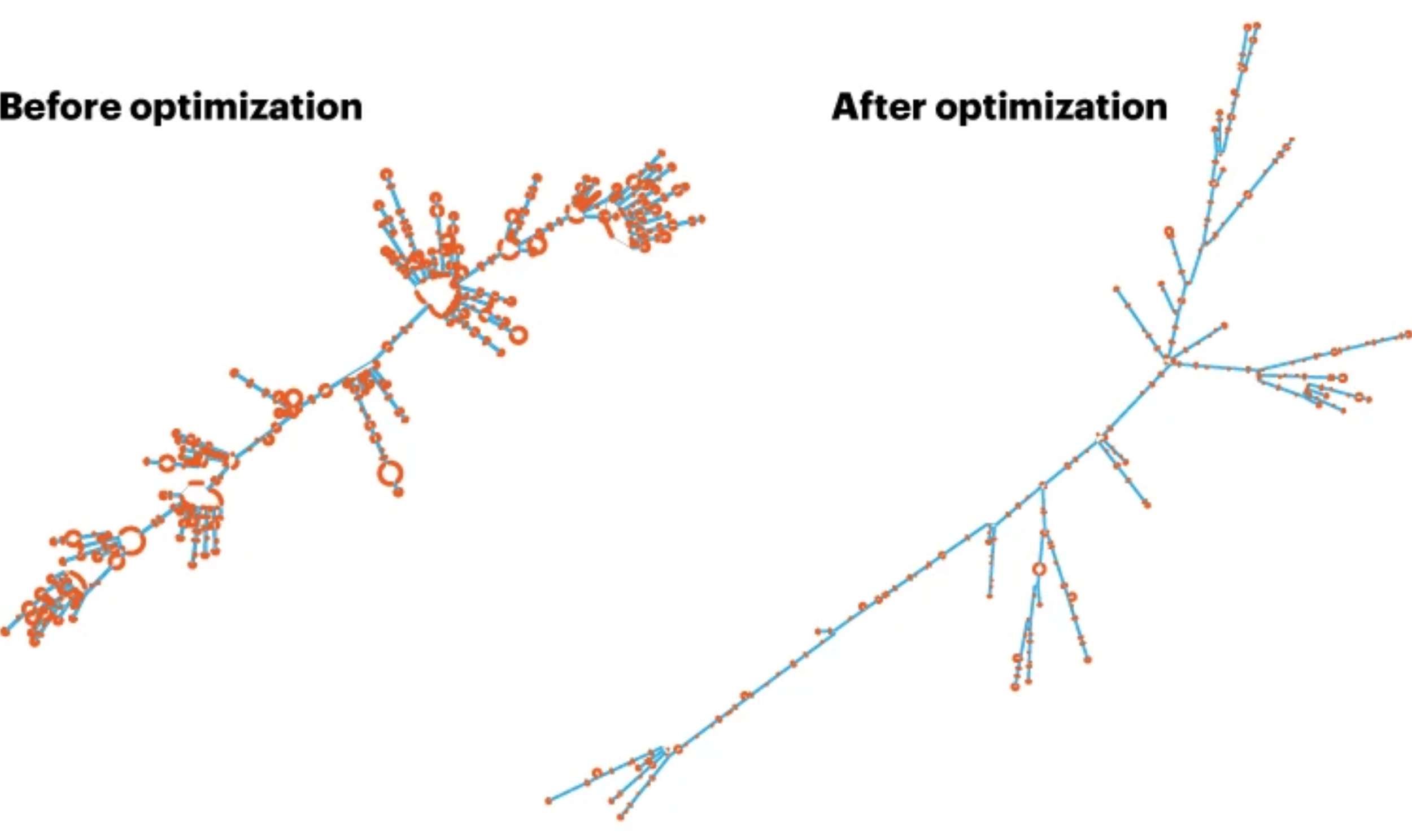
Before optimization, many areas are single-stranded (marked as red) and presumably more prone to degradation. After optimization, most of the single-stranded areas have been replaced with double-stranded structures. Image was obtained and adopted from Nature by Elie Dolgin.
Huang and colleagues demonstrated that by optimizing the mRNA sequence with LinearDesign, the resultant COVID-19 mRNA vaccine shows a longer half-life compared to the Pfizer-BioNTech vaccine. As more intact mRNA was delivered to the cells, LinearDesign-optimized vaccines (sequences A and C) show much higher protein expression than the Pfizer-BioNTech vaccine and the mRNA optimized with the traditional codon optimization approach (sequence H). Moreover, after immunizing mice with two doses of vaccines at an interval of 2 weeks, one of the LinearDesign-optimized vaccines (sequence C) induced stronger antibodies that target the Spike protein and neutralizing antibodies against the SARS-CoV-2 virus.

LinearDesign-optimized vaccines (sequences A and C) show longer half-lives (figure A and B) and higher protein expression (figure C and D)than the Pfizer-BioNTech vaccine and the mRNA optimized with the traditional codon optimization approach (sequence H). Image from Nature by Liang Huang et al.

One of the LinearDesign-optimized vaccines (sequence C) induced stronger antibodies that target (figure A) SARS-CoV-2’s Spike protein and neutralizing antibody against the virus (figure B). Image from Nature by Liang Huang et al.
Although this groundbreaking algorithm tool shows promising results in animal experiments, the safety of the resultant vaccines for use in humans still needs to be proven by clinical trials. As double-stranded RNA is common among viruses, the human immune system is equipped with specialized detectors such as Toll-like receptor 3 (TLR-3) to sense the existence of double-stranded RNAs and alarm the body of a potential viral infection. As the structurally optimized mRNA vaccines have extended double-stranded structures, they may overly activate receptors like TLR-3 and result in more severe side effects.

About the guest author — Chia-Kuei (Simon) Mo
Simon is a Ph.D. candidate in bioinformatics, currently pursuing his studies at Washington University in St. Louis. He completed his bachelor's degree at National Taiwan University and went on to earn a master's degree in Biomedical Engineering from Duke University. Simon's academic pursuits led him to join Dr. Li Ding's lab, where he focuses on investigating cancer biology using a multi-omic approach. Specifically, his current research interest lies in the application of Visium spatial transcriptomics to explore the intricate relationships between tumors and their surrounding microenvironments.